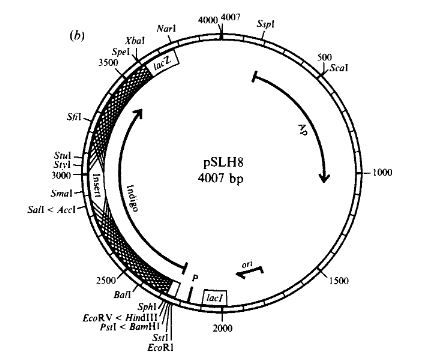
pSHL8 is not the one I have
In my pursuit of an indigo plasmid, I had two options: straight up synthesis, or getting the original plasmid cloned from the rhodococcus gene. Back when I started this project, it would have cost about $600 to get the gene synthesized and stitched together, which would leave very little room for improvement. Fortunately, Dr. Stephen Hart was kind enough to send me the plasmids from his research from 20 years ago.
Unfortunately, the only plasmid that was still intact was labeled pSLH1. pSLH1 is described the paper “Construction of an insertional-inactivation cloning vector for Escherichia coli using a Rhodococcus gene for indigo production” as “The prototype insertional-inactivation cloning vector pSLH1… level of production appeared to be lower than that of other pigment-producing strains…cells produced only slight amounts of pigment even after several days incubation at room temperature.” According to my observations, this is accurate- it takes a while for the transformed bacteria to produce indigo. This seems to be because between the the indigo production gene and the promoter (an in-frame lacZ’a fragment upstream of the gene), there is a stop codon and two rare codons which theoretically prevent the gene from being expressed.
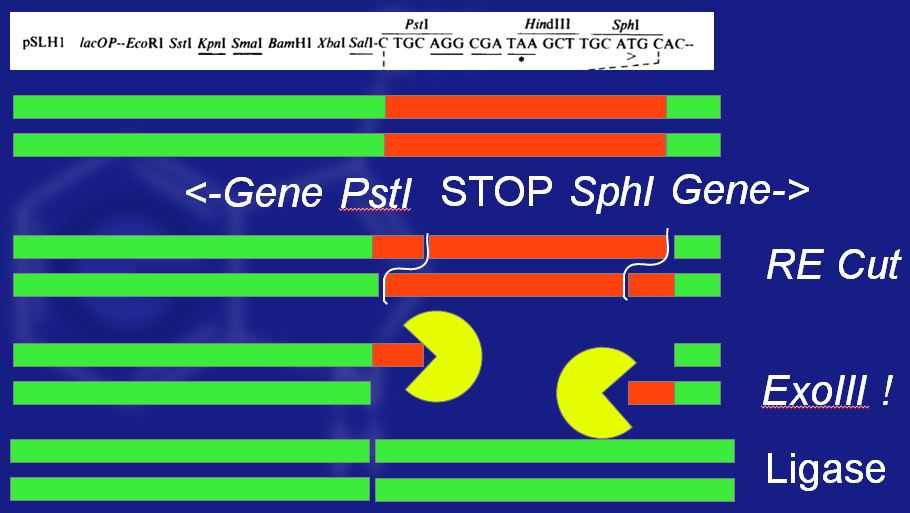
Deletion of stop codon
Hart and Woods went ahead and deleted the deleterious fragment of the gene by cutting out the undesirable DNA, chewing the ends of the DNA back with exonuclease III, and then blunt-end ligating the ends together. Amazingly, this deletion keeps everything in-frame, and produces more indigo than without the deletion. The process is shown in the image above. First, the gene is cut, then the exoIII blunts the ends, and finally they are re-ligated.
In theory, this is great, and it totally works. In practice, something was mislabeled or possibly only a mutated version made it to me 20 years later, because the sequence I got was not the sequence from the paper. It was pretty close, but definitely different. Lets compare:
Modern read -G TCG ACGG
From paper -C TGC AGGC GA
Bolded letters show the “wrong” codons for the cut recognition site (CTGCAG) for PstI. My hypothesis is that the new read is correct, and that the old read was flawed in one way or another. This is because the new read was duplicated exactly the same across 8 independent samples, and the old read was probably done once. The new reads were done on a modern sequencing machine, while the old read was done from radioactive sanger sequencing, which can be hard to interpret, leading to easily transposed letters in the sequence.
The authors may have gotten lucky with the enzyme as well. I was using a recombinant “HF” enzyme, which stands for high fidelity. This means that it has lower star/non-specific activity, reducing the chance it will make an improper cut. To give you an idea of how much more specific the HF version is, I took this snippet from the NEB website:
“The minimum number of units of PstI-HF required to produce star activity in the recommended NEBuffer 4 is (4000 units). The minimum number of units of PstI required to produce star activity in the recommended NEBuffer 3 is (120 units).The minimum number of units of PstI required to produce star activity in NEBuffer 4 is (8 units)”
So the short version is that the HF version is something like 20-500 times more specific. 20 years ago, the PstI may have been even less specific, and allowed for cuts like the one that was made. Or, somewhere along the line, a lossy polymerase copied the DNA wrong and my copy is mutated.
Either way, what actually seemed to happen was that the double-digest with PstI and SphI turned out to be a single-digest. If you look at the actual sequence:
CTTGCATGC
it would be cut like this:
CTTGCATG || C
then blunted to this:
CCTG C
and then ligated to this
CCTGC –> gene
Instead of increasing yield, this introduces a frame-shift mutation, which makes the gene totally useless since it is being translated out of frame. Transforming the DNA yielded ampicilin resistant colonies with no indigo production ability.
So that was a big waste of material. However, there is some good news! The price of DNA synthesis has dropped, and the length of DNA that can be purchased has increased! In these new and exciting times, the cost of synthesizing the gene is only about $200! So I think my next steps are to codon-optimize the gene for e. coli, add restriction sites, then to add a promoter to it and stick the whole thing into p2kb to share with people.

This is a great project – kudos for trying it. Hope it succeeds. I ran the indigo gene (M55641.1) thru the IDT codon optimizer for E.coli, here’s the least complex result, perhaps you’ve already done that….
best of luck
ATG GAC ATC ACT CGC ACC GAG CTG ATG GAC CGT GTT CAC GCT CTG GTA CCA GCG TTT GCG GAG CGT GCG CAG AAA ACG GAG GAG AAC CGC GCC CCC TTG GAC GAG ACG ATC ACT GAC CTG ATC GAT TCA GGT ATT TTG GCG ACC CTT ACC CCC AAG GAG TAT GGG GGC CTG GAA CTG GGT TTG GAT GTT GCA GCG GAT ATT GTT CGG ACG ATC AGT GCT GTG TGC CCT AGC ACC GGT TGG GTC ACC TCC TTC TAC ATT GGT GCC GCG TGG CGG GTG AAT ATC TTT ACC GAG CAG GCT CAA CGC GAG GTG TTT GCG GAC AAA CCA TAC ACG CTG ACC GCC GGC ACC GCT GCA CCG TTG GGA CAG GTC CAA AAA GTG GAT GGT GGG TAT CGG ATT ACG GGC CAG ACG GCG TGG AAC TCG GGC AGT GTT CAC GCG GAG TGG TTT ACC TTC GCG GGT GTT GTG TTT GAA GAG GGT TCC GCC CCG ACA CCG CTT TGG TTT CTG GTG CCG CGC GAG GAC GTC AAA GTG CTG GAT ACC TGG TAC ATT GCG GGG ATG TCA GGC ACC GGC TCC AAC GAT ATT AGC GTC GAT GAT GTG TTC GTT CCA GAA TAT CGT ACA GGC CCA TTT GCC CTG GCC TTA GCT GGA ACC GCC CCG GGC CAA CTG ATT CAC CCG AAC CCC ATG TAT CAT CTG CCG TTC CTG CCT TTT GCA ATG GCG GAA GTT ACG CCA GTG GTG GTT GGT GCG CTG CGT GGC GCT GCG GAT GCC TTC GTT CAA CGT ACC AAA GAC CGC CAA GGC ACA ATC TCC CAA GAG AAA GCA AGC GGC AAA CAG GCT GCG CAG ATG CGC TTG GGC CGG GCA TTG GCG GCA GCT GAC GCG GCG GAA ACA TTA TTA GAT GCC TTT TTT GAA CGT CTG ACC GCT CAG CGG CCC GAG CAG TCC GAC CCA CGG GAC CGC GCA GAA ATG AAG CTC AAA GCT GCA TAT CTT GCG GAT CTG GCC CGC AAC GCC CTC AAT GAT ATG GTG CGT GGT ATC GGT GGT GAC GGC TAC CGG AAC TCC GCG CCG ATT CAG CGC TAT TTC CGT GAC CTG TCT GTC CTG AGT GTG CAC GCC TTC CTG GAC ATT GAC ACC GCG AGT GAG ACC ATT GGC CGT TTT ACC CTT GAT CTC CCG GTA AGC GAT CCG CTT TTA TAA
This is probably my favorite comment, ever. Thanks!