The year is 2013. Why is it when you Google “How do I align my forward and reverse sequences” the two methods that come up are using ClustalW (Clustal Omega), which requires a reference sequence, or some kind of awful witchcraft involving Microsoft word and the search function? I guess there are also some programs out there that you have to pay a large sum to use (squencher etc.) but I certainly don’t have that kind of money.
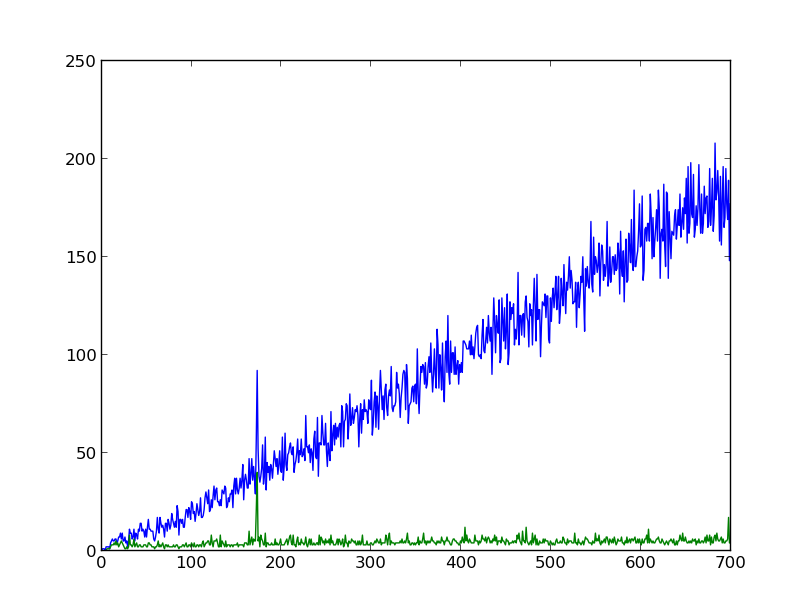
X axis: number of base pairs overlapped from forward and reverse sequence
Blue: number of pairs matching
Green: “longest run”
So I decided to write a little python script to help me do alignments. The output, which you see above, is not very user friendly yet, but it at least gives you a place to start before you devolve to microsoft word (of all things! I will probably use libreoffice though).
Let me explain whats going on up there. The script, which is called DNACrusher, takes the forward and reverse reads from your plasmid, and takes the reverse complement of the reverse read. Then it overlaps the ends that should overlap (the end of the forward read, and the beginning of the reverse complement) by an increasing number of basepairs and counts two things: the longest matching run, and the number of matching base pairs, and stores them in a tuple that remembers how big the overlap is.
The blue line represents the raw number of agreeing basepairs. As you would imagine, as you mash more bp together, the number of agreeing pairs generally go up, as does the noise on that read. I haven’t computed any statistics, but that looks about right. The only anomaly is right before 200 bp overlap, where you have a spike. That turns out to be at 172 bp.
The green line represents the longest run of agreeing bp. This line, as you can see, is relatively flat except for a corresponding peak at 172bp. This makes me fairly certain that there is an overlap of about 172 bp, which seems reasonable. When run against clustalW with the same sequences and a reference sequence, the overlap is about 160 bp, including some gaps in one strand or the other.
You can get the python at ye olde github account. Included the two reads I used to create the graph above. There are only a few methods in the little script, here is a breakdown of how to use them:
Import the DNACrusher module and pyplot
>>>import DNACusher
>>>import matplotlib.pyplot as plt
easy peasyImport your data from a Fasta File
Make a file object from your fasta file
>>>sequence=open(‘Fasta_File.txt’,’r’)
ta-da!
Convert Your File Object to A String
>>>sequence=DNACrusher.fasta_to_string(sequence)
now sequence is a string, which is way easier to deal with
Reverse Complement
>>>sequence=DNACrusher.reverse(sequence)
>>>sequence=DNACrusher.complement(sequence)
‘atgcaata’->’tattgcat’
Overlap N Base Pairs
>>>DNACrusher.crush(forward,reverse,N_BasePairs)
This will return the count of matched bps and the longest run of pbs in a tuple (count, longest)
Overlap Up to N Base Pairs
>>>align=DNACrusher.allign(forward,reverse,N_BasePairs)
This will return a list of tuples [ (pairs_overlapped,(count,longest) , …]
Now to Plot It
>>>plt.plot([ x[1][0] for x in align]) #plot the number of agreement
>>>plt.plot(x[1][1] for x in align]) #plot the record # of runs
>>>plt.show() #show the graph!
Now if you do this with the sequences I provided, you should see the above graph!
>>
